Спінальна м’язова атрофія (СМА) – це рідкісне генетичне захворювання, що зазвичай призводить до ранньої смерті враженої особи. У межах неймовірного дослідження медики вперше продемонстрували ефективність новітнього, але дуже простого методу лікування, завдяки котрому дитина не виявляє жодних ознак захворювання.

СМА є наслідком мутацій гена SMN1, що призводять до дефіциту життєво важливого білка SMN (від англ. survival of motor neuron — «виживання мотонейронів»), який, власне, й відповідає за збереження мотонейронів у спинному мозку. На фоні браку або низького рівня вмісту білка м’язи не отримують сигналів від мозку й поступово атрофуються. Прогресивна атрофія та втрата рухомості насамперед уражає проксимальні м’язи та діафрагму, тому дитина втрачає можливість повзати, ходити, тримати голову, ковтати та дихати. Це захворювання є найбільш поширеною генетичною причиною смерті дітей. Лише у 2016 році в США схвалили використання нусінерсена – першого спеціалізованого препарату, що дозволяє частково компенсувати нестачу білка, а у 2019 році з’явилася перша генетична терапія, що замінює дефектний ген та припиняє прогресування хвороби. Втім, ці методи лікування СМА не є ідеальними, адже вони не можуть запобігти появі симптомів та не завжди дозволяють відновити нормальну функцію організму.

Об’єктом нового дослідження став препарат рісдіплам, що є аналогом нусінерсена, але має велику перевагу: його рецептура передбачає пероральне введення препарату, а не інтратекальне (тобто прямо у хребтовий канал). В умовах традиційної терапії він також сповільнює прогресування захворювання та допомагає покращити моторну функцію й підвищити якість життя дитини. Але, на жаль, його не можна вважати панацеєю. Зазвичай терапія рісдіпламом починається одразу після народження дитини: чим раніше втрутитися, тим кращими будуть результати. То ж дослідники з Дитячої дослідницької лікарні Сейнт Джуд вирішили спробувати розпочати лікування ще до народження дитини.
Обидвоє батьків у парі, що взяла участь у досліджені, були носіями мутації гена SMN1, що підвищувало ризик народження малечі зі СМА. До того ж раніше вони вже тратили дитину через це захворювання у віці 16 місяців. Генетичний пренатальний скринінг показав, що їхня друга дитина також не має копій гена SMN1, отже, найімовірніше, вона народилася б зі спінальною м’язовою атрофією типу I. У межах дослідження протягом останніх шести тижнів вагітності препарат рісдіплам вживала сама матір. Після пологів від віку всього одного тижня лікарі продовжили давати ліки вже малечі. На думку дослідників, дівчинці доведеться пити рісдіплам протягом всього життя, але терапія одразу показала позитивний результат. Так, в її крові міститься вищий рівень вмісту білка SMN, ніж в інших дітей із аналогічною генетичною мутацією. Рівень пошкодження нервів також є значно нижчим, і навіть у віці 30 місяців дівча розвивається нормально, а її м’язи не мають ознак атрофії.
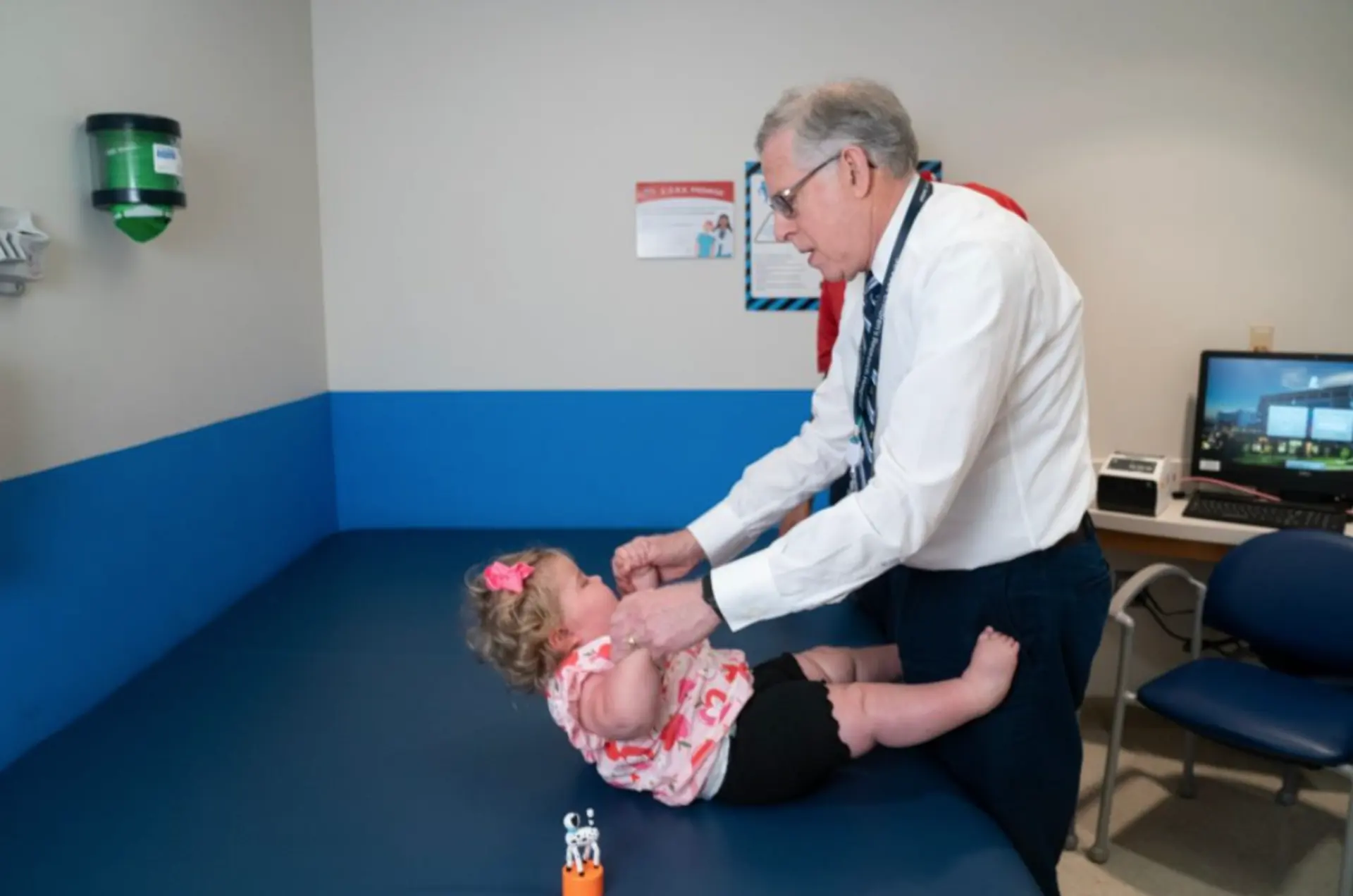
За словами автора дослідження Річарда Фінкеля, його колеги були надзвичайно вражені тим, що завдяки завчасному початку використання рісдіплама дитина не проявляє ознак СМА. Звичайно ж, дослідження за участі всього одного пацієнта не може слугувати доказом ефективності техніки для всієї ураженої популяції, але воно закладає чудову основу для більш масштабного експерименту. На прикладі цієї родини вони переконалися, що як матір, так і дитина добре перенесли терапію, отже, ця ідея є здійсненою, безпечною та вартою подальшого вивчення.